畫作名稱:夢想潛水艇
畫作介紹:我的夢想是成為海洋科學家,雖然無法行走但仍然可乘坐潛水艇,遨遊探索神秘新海洋世界。
作 者:李亮葟
個案故事
我從小患有罕病「脊髓性肌肉萎縮症」,判定為重度身心障礙者,自從1歲多時發病後,全身隨著年齡成長而漸漸退化無力,因而自幼無法行走,手也漸漸舉不高了,目前需要依靠電動輪椅移行代步。
我喜歡創作不論是寫作和繪畫,想傳遞不要因為疾病限制自己的發展,要克服疾病的限制而勇於面對生活,用「創造自己的光」希望能引領他人。
疾病名稱
脊髓性肌肉萎縮症(Spinal Muscular Atrophy, SMA)
疾病介紹
脊髓性肌肉萎縮症是一種遺傳性運動神經元退化疾病,約95%的脊髓性肌肉萎縮症患者致病原因為第五號染色體SMN1基因發生缺失(deletion)或轉換(conversion)突變造成,其餘約5%則伴隨SMN1基因的點突變(point mutation)或空間排列錯置所造成SMN1基因突變。SMN基因突變會導致脊髓的前角細胞(運動神經元)退化,造成肌肉呈對稱性逐漸無力及萎縮,下肢較上肢嚴重且身體近端靠近軀幹的部位較遠端易受到影響,影響患者控制隨意肌肉的能力。發病年齡從出生到成年皆有可能發生,每個人依照發病年齡以及疾病的嚴重程度,症狀會有很大的差異。依文獻報告,各族群帶因率約1/40~1/100。
在台灣,每40人就有一名是帶因者,帶因率僅次於海洋性貧血。由於帶因率和致病率皆高,因此在2008年時美國醫學遺傳學暨基因體學學會 (American College of Medical Genetics and Genomics, ACMG) 已經開始推廣,建議夫妻雙方於婚前或懷孕早期即進行篩檢;而在2017年美國婦產科醫學會 (American College of Obstetricians and Gynecologists, ACOG) 建議脊髓肌肉萎縮症基因帶因檢測必須不分種族的提供給所有婦女。
臨床症狀
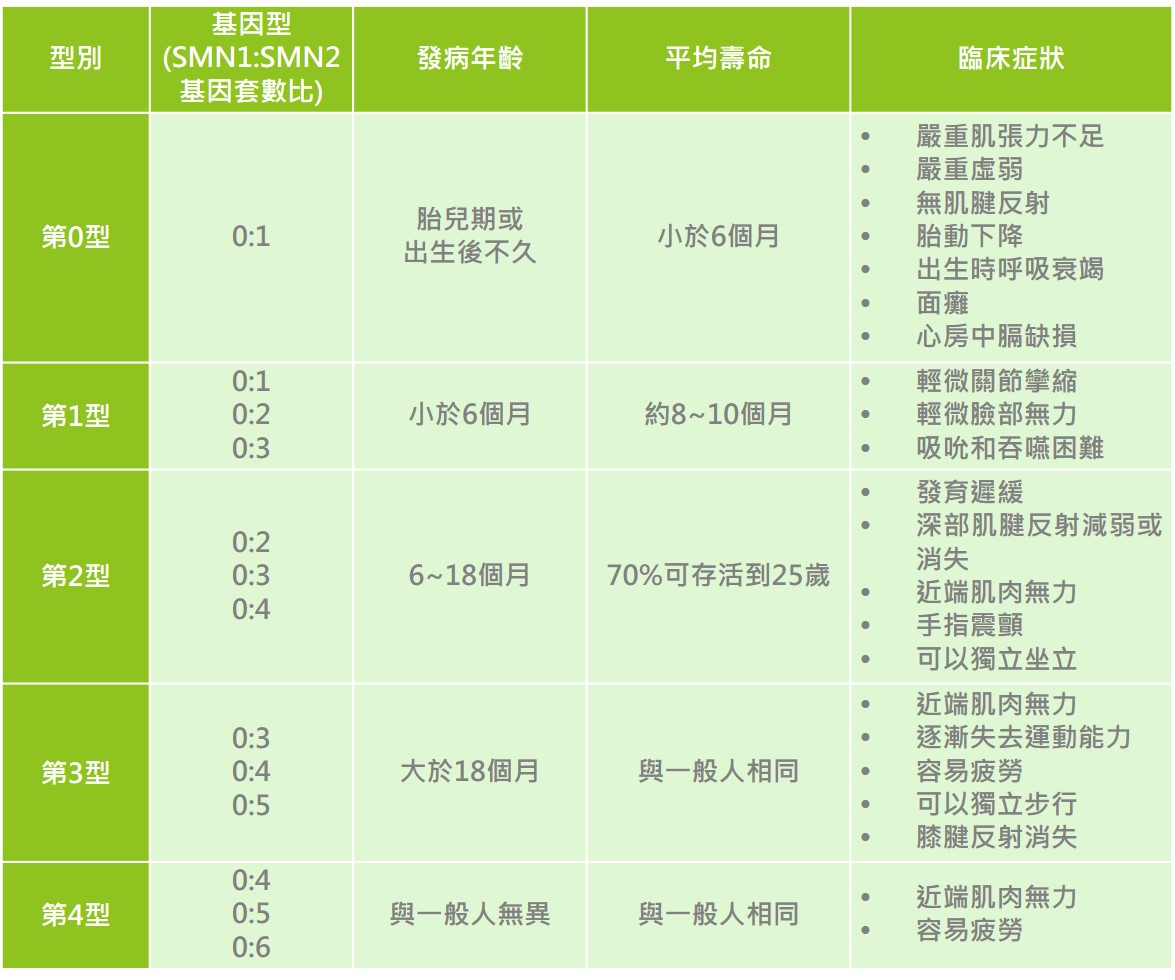
脊髓性肌肉萎縮症依據發病年齡、疾病嚴重度及肌肉受影響程度分為五個型別:
遺傳模式
體染色體隱性遺傳,若父母親雙方皆為同基因突變之帶因者,其本身並不會發病或有任何症狀,則下一代不分性別有 1/4 的機率生下患者。
檢驗方式
臨床診斷會先透過專業醫師的臨床特徵評估,如:動作障礙、近端肌肉無力、肌腱反射降低或喪失等;以往肌肉切片是確定脊髓性肌肉萎縮症最重要的方法,目前可利用分子檢測方式進行SMN基因劑量分析。針對有臨床特徵之患者、有家族疾病史之家族成員,建議可與專業醫師進行遺傳諮詢,透過臨床評估與基因檢測的方式,讓脊髓性肌肉萎縮症患者可以及早診斷與醫療協助。
參考資料
- GeneReviews®
- MedlinePlus Genetics
- 罕見遺傳疾病一點通