畫作名稱:探索基因DNA的奧秘
畫作介紹:希望在未來,能以更多元的方法探索 DNA 的奧秘,揭開罕見疾病背後的秘密,為罕病病友帶來新的曙光。
作 者:黃梅珠
個案故事
梅珠是一位Fabry氏症病友,長年與疾病相伴,讓他對身體裡的「秘密」充滿疑問。這幅《探索基因DNA的奧秘》,DNA隱藏著生命的密碼,也承載著他對未來的希望。雖然疾病帶來許多挑戰,但他相信科學能一步步解開謎題,為病友帶來新的曙光。透過創作,他把對生命的好奇與堅強,化作一幅充滿勇氣與夢想的作品。
疾病名稱
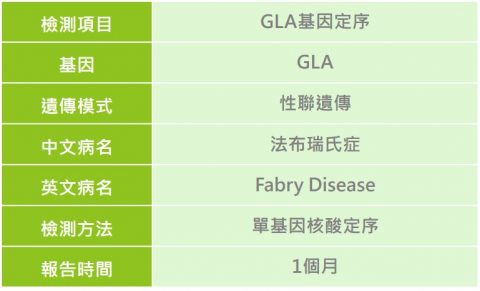
法布瑞氏症(Fabry Disease)
疾病介紹
法布瑞氏症是一種罕見的遺傳性溶小體儲積症(Lysosomal Storage Disorders, LSDs),致病原因為X染色體上的GLA基因變異所致,此基因主要負責製造α-galactosidase(簡稱α-GAL)酵素。當體內缺乏此酵素時,會使得醣脂質,特別是globotriaosylceramide(簡稱GL-3或Gb-3)無法被代謝,於是容易堆積在腎絲球、腎小管的上皮細胞、心肌細胞與瓣膜纖維細胞,造成腎臟、心血管及腦血管病變,此外醣脂質的堆積也會造成周邊神經病變,引起四肢劇烈疼痛。
臨床症狀
通常於兒童與青少年時期開始出現臨床表徵,典型的法布瑞氏症患者肢體末端會出現間歇性的疼痛、燒灼痛感及感覺異常;皮膚上會出現紫黑色的皮膚病變,稱為血管角質瘤,或是表皮出現紅疹;眼睛的角膜呈現輻射狀或螺旋狀濁斑。在成人時期出現進行性的腎臟、心血管及腦血管病變,最後進展為腎臟衰竭、心臟合併症、早發性中風,成為威脅生命的主因,也有部份的患者可能會有腸胃道不適及聽力損失等症狀。台灣多數患者屬於非典型法布瑞氏症,症狀以心臟症狀為主,缺乏典型法布瑞氏症的臨床症狀,在幼年、青少年期症狀不明顯,通常於40~50歲左右臨床症狀才逐漸明顯。
遺傳模式
性聯遺傳。女性若帶有一個GLA基因突變點位,應為帶因者,但由於X染色體去活化機制,亦有可能出現輕微或具有多變性的臨床表徵;男性則因僅有一條X染色體,故只要帶有此基因突變點位,即為患者。
檢驗方式
法布瑞氏症為新生兒篩檢自選項目之一,測定濾紙血片檢體中α-GAL的酵素數值,若低於正常值,需進行複檢,若複檢結果仍為異常時,需至各大醫療中心進行確認診斷。
除了專業醫師的臨床評估及詢問家族史外,可透過實驗室檢測酵素活性及尿液GL-3分析等一系列檢查進行確認診斷,而對於女性帶因者,檢測酵素的活性有其侷限性,因為可能症狀輕微,酵素活性仍在正常範圍內,需透過分子基因檢測方式進行確認診斷。
針對有臨床特徵之患者、有家族疾病史之家族成員等,建議可與專業醫師進行遺傳諮詢,透過臨床評估與基因檢測的方式,讓法布瑞氏症之患者可以及早診斷治療與醫療協助。
參考資料
- GeneReviews®
- StatPearls
- 罕見疾病基金會