

疾病簡介
體染色體隱性遺傳多囊性腎臟病(Autosomal recessive polycystic kidney disease, ARPKD),致病原因為位於第6號染色體上的PKHD1基因突變,影響形成纖毛結構相關的纖維囊蛋白的功能,造成嬰兒或幼童的肝臟及腎臟出現囊泡與纖維化特徵。肝臟的表現常有肝門靜脈周圍的纖維化,膽管的增生或肝門靜脈高壓等症狀。腎臟的侵犯主要是有許多微小的囊泡所造成,這些囊泡的產生主要是由於腎元的集尿管不正常的擴張所造成,而腎功能受損的程度與腎元受囊泡侵犯的程度相關。
PKHD1基因帶因率約為1~2%。
臨床症狀
患者可能於胎兒時期即可發現腎臟異常腫大或多囊腎等症狀,造成羊水過少而導致肺部發育不全、獨特的面部特徵,如小下巴、低位耳等;在出生時有可能因呼吸困難引起呼吸衰竭,或感染肺炎而死亡。患者臨床表現變異性大,隨著年齡的增長主要的併發症有腎功能衰竭、電解質失衡與高血壓,還有肝臟方面的異常,如肝腫大、膽管擴張、肝門靜脈高壓等症狀。
目前以支持性治療為主,呼吸困難的患者使用呼吸器以輔助呼吸,控制血壓避免發生高血壓,維持電解質平衡及腎臟管理等,若發生腎衰竭,需進做血液透析或進行腎臟移植。
遺傳模式
體染色體隱性遺傳,若父母親雙方皆為同基因突變之帶因者,其本身並不會發病或有任何症狀,則下一代不分性別有 1/4 的機率生下患者。
檢驗方式
由專業臨床醫師評估患者的臨床症狀、家族史、超音波等檢查項目。胎兒或新生兒時期,透過腎臟超音波檢查患者通常有雙側腎臟腫大及皮質髓質分化不良,並伴有迴聲增加、囊腫及羊水過少等症狀,肝臟超音波有膽管擴張、肝門靜脈異常等,以及進行組織切片確認肝纖維化。
針對有臨床特徵之患者、有家族疾病史之家族成員等,建議可與專業醫師進行遺傳諮詢,透過臨床評估與基因檢測的方式,讓體染色體隱性遺傳多囊性腎臟病之患者可以及早診斷治療與醫療協助。
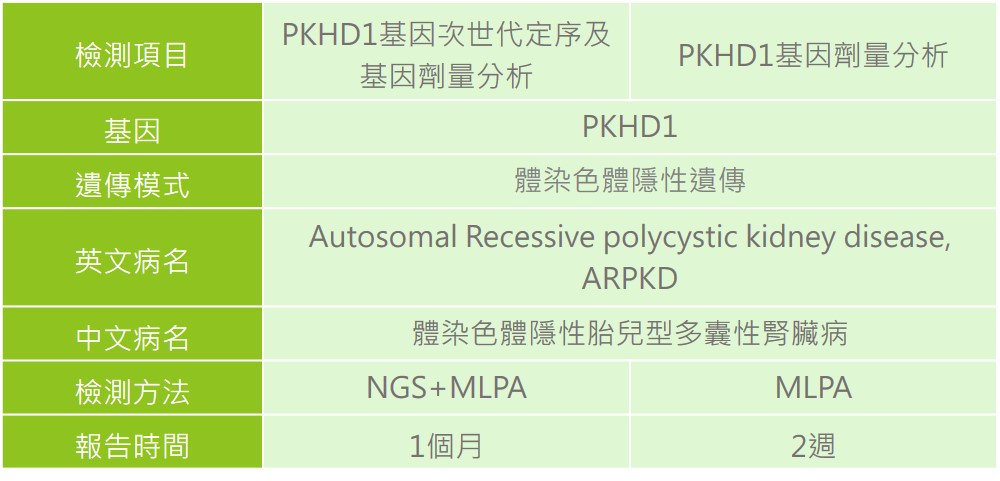
首選慧智(相關檢測如需進一步諮詢,請洽詢專業醫療人員或醫師)
參考資料
- Hum Mutat. 2004;23(5):453-63.
- GeneReviews®
- 罕見疾病一點通
- Orphanet



