

畫作名稱:罕見劇場
畫作介紹:媽媽帶我去看歌仔戲,演員們身穿華服、唱腔細膩、身段柔軟,非常厲害。
作者名稱:何冠毅
個案故事
冠毅今年12歲,個性活潑開朗,「愛伯特氏症」的我至今已經動了約20次手術,我的外觀和別人不一樣,但我沒有放棄自己,除了寫字、畫畫、玩電腦,我也參與很多活動:打棒球、踢足球、唱歌、音樂律動等,讓自己的生活多彩多姿,享受生活。
疾病名稱
愛伯特氏症 (Apert Syndrome)
疾病介紹
愛伯特氏症是一種顱顏發育不良症,致病原因為第10號染色體上的FGFR2基因變異所致,主要病徵是由於顱骨縫過早閉合而導致顱顏畸形,並伴隨四肢及多部位骨骼畸形,合併症狀從皮膚、軟組織到骨頭都可能發生。愛伯特氏症的發生率約在1/65,000〜1/88,000。
臨床症狀
患者因顱骨縫過早閉合,所以一出生即有症狀,如:臉中央發育不全、眼球突出、顎裂、咬合不正、鼻子較短且顱骨高又扁平、額鼻交界處較為凹陷,並且常見伴隨手指指節間關節緊連、手腳併指、拇指較為寬大等。因臉部的凹陷導致鼻咽空間減少,鼻呼吸道因而阻塞,患者常使用口呼吸,易造成睡眠呼吸中止,提升口腔畸形及牙齒的問題。
部分患者在其他身體系統或骨骼方面也會出現症狀,像是傳導性聽力障礙、多汗症、有嚴重面皰的油性皮膚及骨關節異常等問題,少見的症狀為先天性心臟結構異常、腸胃道或泌尿道結構異常,大部分患者的智能為正常或輕微障礙。
主要治療方式是以整形外科手術開刀為主,來減輕症狀的嚴重度。一般而言,建議於出生後3個月至1歲間進行前額輪廓的矯正手術,透過及早矯正顱骨異常,可降低因顱縫過早閉合而限制頭骨生長,造成腦室擴大或水腦的風險。因患者的臨床症狀有個體差異性,故手術治療需多個專科醫療團隊合作進行。
遺傳模式
愛伯特氏症為體染色體顯性之遺傳模式,具有一個對偶基因帶有異常點位突變,即有可能會致病;若父母任一方為患者,則下一代不分性別有1/2機率為患者。多數患者的父母雙方皆為正常人,是在胚胎發育過程中,因自發性的基因變異而導致罹患此病。
檢驗方式
1. 針對有臨床特徵的患者或有家族疾病史的家族成員,建議可與專業醫師進行遺傳諮詢,透過臨床評估,如外觀檢查並輔以顱骨、脊椎、手部及腳部放射線檢查、頭部電腦斷層攝影與腦部核磁共振來確認顱縫早閉及骨骼異常,新生兒聽力篩檢確認是否有聽力障礙問題,還有基因檢測的方式,讓愛伯特氏症患者可以及早診斷治療與醫療協助。
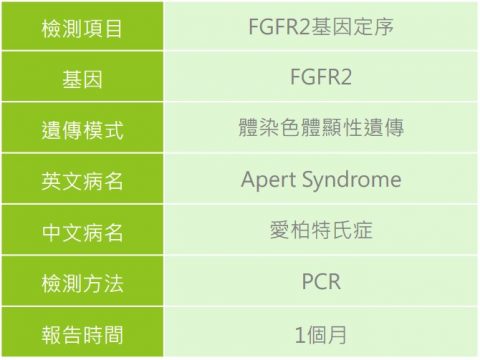
2.確認胎兒是否為愛柏特氏症,於產前可選擇進行染色體相關檢測像是非侵染色體篩檢(NIPS)或侵入性檢測以絨毛膜或羊水檢體進行全方位複合式晶片檢查(僅針對FGFR2之特定好發點位分析),確認胎兒是否為愛柏特氏症患者,檢測前需與您的主治醫師進行評估與討論。

參考資料
- 罕見疾病一點通
- GeneReviews®
- National Institutes of Health



