

透過基因檢測,我們可以為孩子做的更多更好!!!
可以預知疾病的發生,若不幸帶有致病變異,透過早期進行預防及介入治療,為寶寶健康把關,也是給予寶寶最棒的第一份禮物。
個案小故事 (本篇故事經過重新編寫,非特定案例)
回想起半年前,霏霏媽媽和其他新手媽媽一樣,在產前進行了很多項檢查結果都正常,且生產過程也非常地順利,但在霏霏出生後一週,霏霏媽媽接到院所的來電,通知小朋友在新生兒篩檢有一項數值異常,是與肝醣代謝有關的龐貝氏症,確認霏霏診斷的醫生告知龐貝氏症患者需使用酵素替代藥物降低後續併發症的發生;在治療期間,霏霏媽媽也收到出生時自費幫霏霏進行的新生兒基因檢測報告,報告結果在龐貝氏症相關基因上有找到致病位置,同時也提供父母親為龐貝氏症帶因者可能性及再發生率的風險。
霏霏媽媽不禁感嘆著,原來不是產前超音波及檢查都正常就沒事,也不是她跟先生都正常小孩就不會得到遺傳性疾病,看著治療後的霏霏發展與一般孩子無異,已經會撐起身體探索著世界,萬分慶幸現在的醫學技術發達,可以及早診斷及早治療,讓這些罕病小孩可以健康的成長。
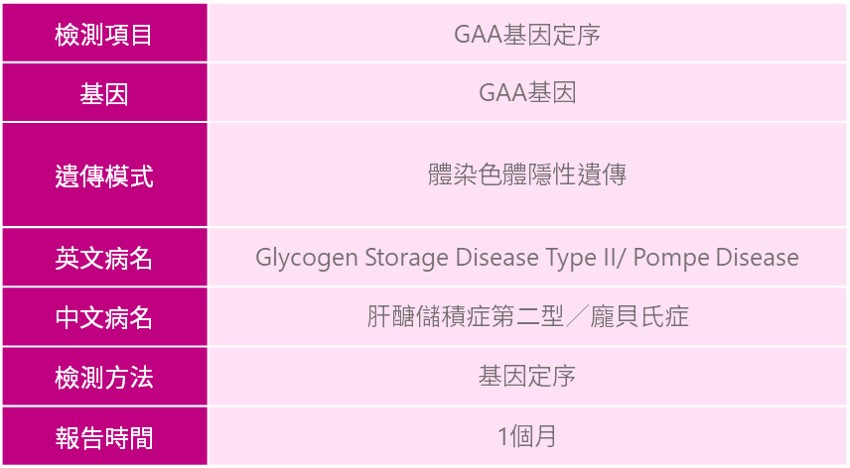
肝醣儲積症第二型(Glycogen Storage Disease Type II)
龐貝氏症(Pompe Disease)
疾病簡介
肝醣儲積症(Glycogen Storage Disease)是罕見的遺傳代謝疾病,主要是患者體內無法正常代謝肝醣所致,這些肝醣會大量堆積在器官或組織影響正常運作,尤其在肌肉組織中影響更為嚴重。在臨床上目前可區分13大類,因篇幅有限本篇以介紹肝醣儲積症第二型為主。
肝醣儲積症第二型(Glycogen Storage Disease type II)又稱龐貝氏症(Pompe Disease),是源至-於1932年荷蘭醫師J.C Pompe診斷出患病孩童,故採用此命名。龐貝氏症致病原因為位於第17號染色體上的GAA基因發生突變,導致溶小體中acid alpha-glucosidase(GAA)酵素缺乏,使得肝醣無法有效轉換成葡萄糖造成肝醣逐漸堆積,進而傷害肌肉功能。大部分患者會出現漸進性的肌肉無力,當不同部位的肌肉功能逐漸退化時,則有可能會有全身性臨床表徵出現,尤其以心臟和呼吸道系統最為嚴重。龐貝氏症在全球發生率約1/40,000,在不同種族中會有差異;根據統計在台灣,嬰兒型發生率為1/40,000,晚發型發生率為1/20,000。
臨床症狀
患者最主要臨床症狀為肌肉相關問題,以肌肉張力降低、無力為主。龐貝氏症依照發病時間點可分為嬰兒型和晚發型兩種,其中1/3患者為嬰兒型。
嬰兒型龐貝氏症
患者其GAA酵素活性通常<1%,發病時間約在出生後幾個月內開始,可能會有嚴重肌肉無力、舌頭肥大、心臟肥大、肝臟肥大、呼吸困難、無法如期達到發育標的等臨床表徵;若不治療,患者通常在一歲內會因心力衰竭而死亡。
晚發型龐貝氏症
患者通常還保留部分GAA酵素活性,發病時間從兒童時期到晚年都有可能發生,一開始的症狀與其他肌肉疾病症狀相類似,故常會延誤診斷。因個體差異性故臨床症狀變化較大,病人可能輕微到只有肌肉無力等症狀,嚴重則可能需要使用輪椅或呼吸器輔助。通常發病時間越早,其臨床症狀及惡化程度較為嚴重。
過去只能以支持性療法治療,目前有酵素替代療法(Enzyme replacement therapy, ERT),且於2005年核可列為罕病用藥並納入健保給付,此藥物-Myozyme是補充病人缺乏的GAA酵素,患者需每兩周由靜脈注射一次,及早發現及早治療,可有效延長患者生命及改善生活品質。
遺傳模式
龐貝氏症(Pompe Disease)遺傳模式為體染色體隱性遺傳,通常夫妻雙方為此疾病之帶因者,則下一代有1/4的機率生下患病之胎兒,3/4的機率生下正常之胎兒。
檢驗方式
龐貝氏症(Pompe Disease)可透過新生兒篩檢的方式,測定血片中acid alpha-glucosidase(GAA)酵素活性,檢測出嬰兒型患者,若濃度低於正常值,將接獲通知並依照指示回診,專業醫師會進行臨床特徵評估、生化血液檢測、影像等進行疾病診斷;另外針對有臨床特徵之患者、家族疾病史之家族成員,建議可與專業醫師進行遺傳諮詢,透過臨床評估與基因檢測的方式,讓龐貝氏症患者可以及早診斷治療與醫療協助。
首選慧智 (相關檢測如需進一步諮詢,請洽詢專業醫療人員或醫師)
參考資料
GeneReviews®
財團法人罕見疾病基金會



