

疾病簡介
裘馨氏肌肉萎縮症(Duchenne muscular dystrophy,DMD)是肌肉萎縮症中最常見的一種,根據歐美統計每十萬新生男嬰中有約20~30人患病,其致病原因是位於X染色體短臂Xp21的Dystrophin基因(DMD gene)有異常而造成的,DMD基因主要控制肌肉組織中肌縮蛋白(dystrophin)的合成,若此基因有異常或突變就會影響蛋白質的合成,進而導致骨骼肌肉細胞壞死而致病。帶有此基因異常者會導致本身肌肉組織隨著年紀衰退外,於晚期也可能因控制呼吸與行動的肌肉萎縮,而導致其他併發症甚至有死亡的風險。
臨床症狀
患者剛出生時發育大致正常且無明顯症狀,隨著年紀增長肌肉組織會逐漸衰退,初期病徵出現於嬰幼兒(約3~7歲)時期,父母親會開始注意到動作發展的時程會較一般孩子緩慢,像是學坐、站、走等基礎行為,且患者走路時容易跌倒,墊腳尖、跑步、上下樓梯的等動作較笨拙與吃力。初期僅於下肢出現肌無力,全身肌肉以骨盆近端的大肌肉先呈現左右對稱的影響;由於腰部脊椎旁的肌肉也受影響,故患者的脊椎前後異常彎曲,走路時小腹往前凸,且因臀部肌肉無力,身體會左右搖晃和肩膀後縮的方式來代償,跌倒時不易自然地站起,而需用雙手按在伸展的膝部,然後按在大腿上慢慢由腿部「爬」上來,以取得直立的姿勢稱為「高爾移動」(Gower manoeuvre)。在孩童時期(約12~13歲)就無法自行走路需靠支架輔具或輪椅代步,因患者會有多處關節攣縮與變形,在疾病後期可能因呼吸肌群或心臟肌肉受到波及,會有呼吸困難之臨床症狀,需要使用呼吸器輔助,因此患者常會因心肺衰竭或感染而走向生命終點。
遺傳模式
裘馨氏肌肉萎縮症為性聯隱性遺傳模式(X-linked recessive),若母親的X染色體其中一條帶有此基因異常,則每胎皆有1/2機率將帶有基因異常的X染色體遺傳給下一代;若胎兒遺傳到帶有基因異常的X染色體,女性通常不會有臨床症狀並稱之為帶因者,而男性則為患者。
檢驗方式
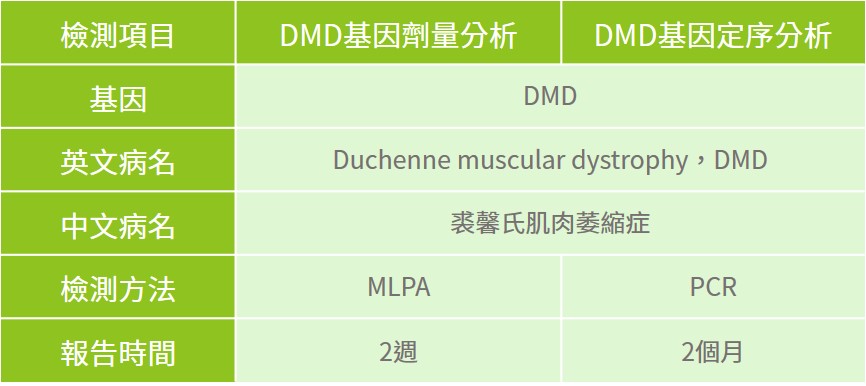
目前可透過專業醫師評估後進行酵素檢測、肌電圖、肌肉切片及心電圖等影像學來做為疾病診斷,此外也可以利用分子檢測方式進行DMD基因檢測;針對有臨床特徵之患者、有家族疾病史之家族成員等,建議可與專業醫師進行遺傳諮詢,透過臨床評估與基因檢測的方式,讓裘馨氏肌肉萎縮症患者可以及早診斷治療與醫療協助。
首選慧智 (相關檢測如需進一步諮詢,請洽詢專業醫療人員或醫師)
參考資料
- GeneReviews®
- 財團法人罕見疾病基金會
- 罕見遺傳疾病一點通
- 中華民國肌萎縮症病友協會



