

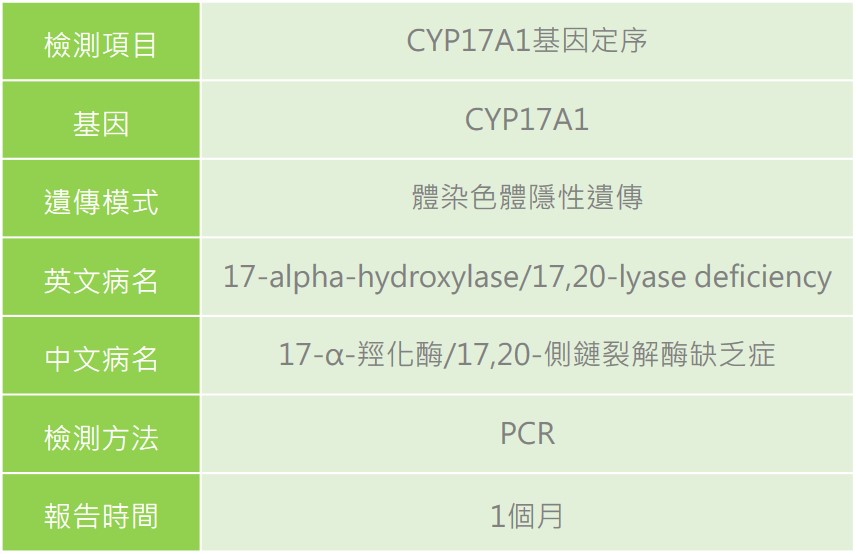
疾病簡介
17-α-羥化酶缺乏引起之先天性腎上腺增生症(Congenital adrenal hyperplasia (CAH) due to 17-alpha-hydroxylase deficiency),致病原因為位於第10號染色體上的CYP17A1基因突變所致,使得參與合成腎上腺皮質激素的酵素(17-α-羥化酶)缺乏,該酵素缺乏將導致皮質醇(cortisol)、雄激素(androgen)缺失及促腎上腺皮質素(adrenocorticotropic hormone)生成過量。依酵素缺乏程度會有高血壓、低血鉀和性特徵發育異常的情形。
臨床症狀
多數患者可能於青春期或是成年初期才被診斷,常見的臨床症狀如:高血壓和低血鉀;外觀的表現特徵根據酵素缺乏的多寡和性別有關,女性患者會有青春期延遲、原發性無月經、缺乏第二性徵等症狀;男性患者同樣會有青春期延遲、第二性徵不明顯,甚至於出生時具有典型的女性外生殖器的問題。不論男性或女性患者皆會有不孕的問題。
遺傳模式
體染色體隱性遺傳,若父母親雙方皆為同基因突變之帶因者,其本身並不會發病或有任何症狀,則下一代不分性別有1/4的機率生下患者。
檢驗方式
先天性腎上腺增生症為新生兒篩檢檢查項目之一,測定濾紙血片檢體中17-OHP的含量,若濃度高於正常值,需進行複檢,若複檢結果仍為異常時,需至各大醫療中心進行確認診斷。
除了由專業醫師進行臨床症狀評估之外,血液染色體檢查、檢測血液的pH值、血液中鉀以及鈉離子的濃度、尿液中孕三醇(Pregnanetriol)及17-酮類固醇(17-ketosteroid)濃度等可以協助醫師診斷。
CYP17A1基因變異佔所有先天性腎上腺增生症患者約1%左右,有臨床特徵之患者、有家族疾病史之家族成員等,建議可與專業醫師進行遺傳諮詢,必要時可使用分子檢測方式進行基因檢測,早期診斷並治療及追蹤,通常預後較為良好。
首選慧智 (相關檢測如需進一步諮詢,請洽詢專業醫療人員或醫師)
參考資料
- Discov Med. 2017;24(133):175-182.
- Fertil Steril. 2014;101(2):317-322
- J Biol Chem. 1991;266(24):15992-15998.
- StatPearls



