

疾病簡介
苯酮尿症(Phenylketonuria, PKU) 是一種代謝異常的遺傳性疾病,發生率約為1/40,000,因致病機轉不同,可分為兩大類別。
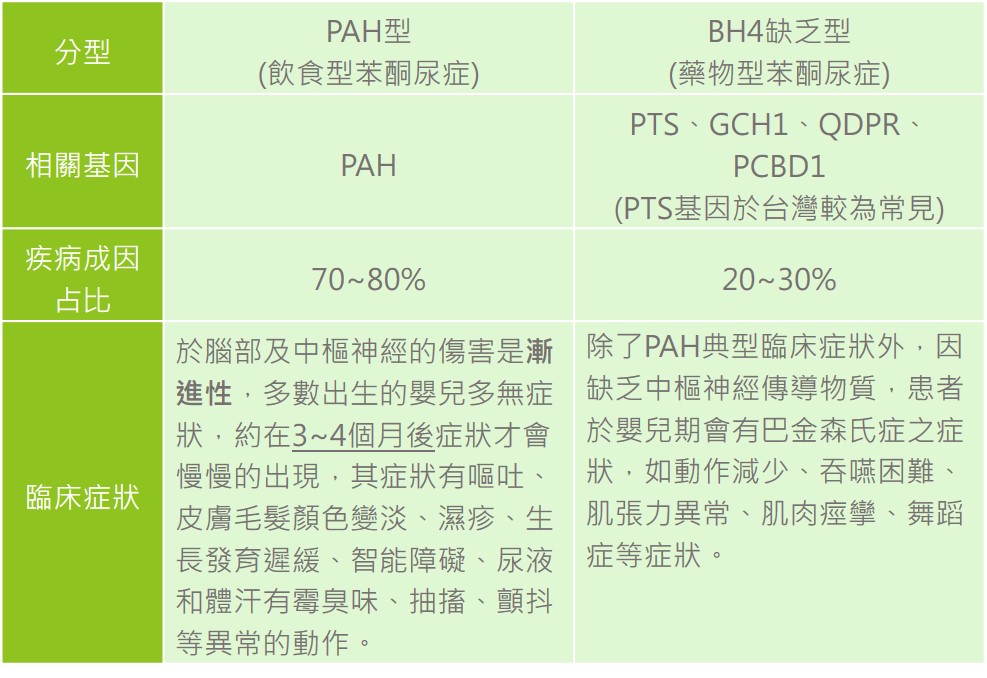
第一類為苯丙胺酸羥化酶缺乏型(PAH型),亦為飲食型苯酮尿症,致病原因為位於第12號染色體上的PAH基因發生突變導致酵素失去功能,造成苯丙胺酸堆積無法正常代謝,在體內大量堆積有毒的代謝產物,造成神經傷害;在尿液中也會檢測出過量的Phenylpyruvate、Phenylacetate、Phenyllactate等物質,使得尿液和身體會出現腐臭味。
第二類為BH4缺乏型,又名為藥物型苯酮尿症,已知PTS、GCH1、QDPR、PCBD1等相關基因與BH4缺乏型有關,在台灣最常見的是PTS基因突變,導致無法產生足量之BH4協助PAH酵素代謝苯丙胺酸,造成有毒代謝產物大量堆積;另外,BH4也是大腦中產生神經傳導物質的重要輔酶之一,當缺乏BH4會導致中樞神經傳導物質Dopa和5-OH Tryptophan無法被合成,進而出現神經學相關症狀。
臨床症狀
遺傳模式
體染色體隱性遺傳,若父母親雙方皆為同基因突變之帶因者,其本身並不會發病或有任何症狀,則下一代不分性別有1/4的機率生下患者。
檢驗方式
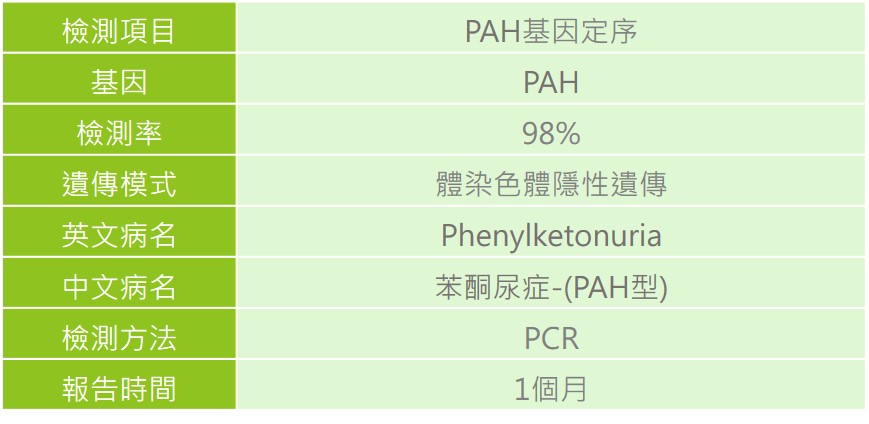
苯酮尿症為新生兒篩檢檢測項目之一,測定濾紙血片檢體中苯丙胺酸之含量,若濃度高於正常值,需進行複檢,若複檢結果仍為異常時,需至各大醫療中心進行確認診斷。除了專業醫師的評估外,透過實驗室檢測血液中苯丙胺酸及酪胺酸的濃度、尿液有機酸氣相層析質譜分析(GC/Mass)及尿液高效液相層析定量新喋呤(Neopterin)和生物喋呤(Biopterin)、紅血球DHPR活性定量及BH4輔酶的負荷試驗(BH4 loading test)等檢驗,依據檢測結果評估是否為苯酮尿症患者及疾病分型。另外,也可透過基因檢測方式進行確認診斷。
首選慧智(相關檢測如需進一步諮詢,請洽詢專業醫療人員或醫師)
參考資料
- Clin Chem. 1993;39(11 Pt 1):2354-5.
- GeneReviews®
- National Institutes of Health
- 罕見疾病一點通



